Genetic Counseling
Couples with sickle cell trait can receive counseling to understand the risk of passing SCD to their children.
A comprehensive guide to causes, symptoms, treatment, and living with SCD
Learn MoreSickle cell disease (SCD) is a serious, inherited blood disorder that affects millions worldwide. This page provides a comprehensive overview of SCD, including its causes, symptoms, treatments, and global impact, to raise awareness and support those affected.
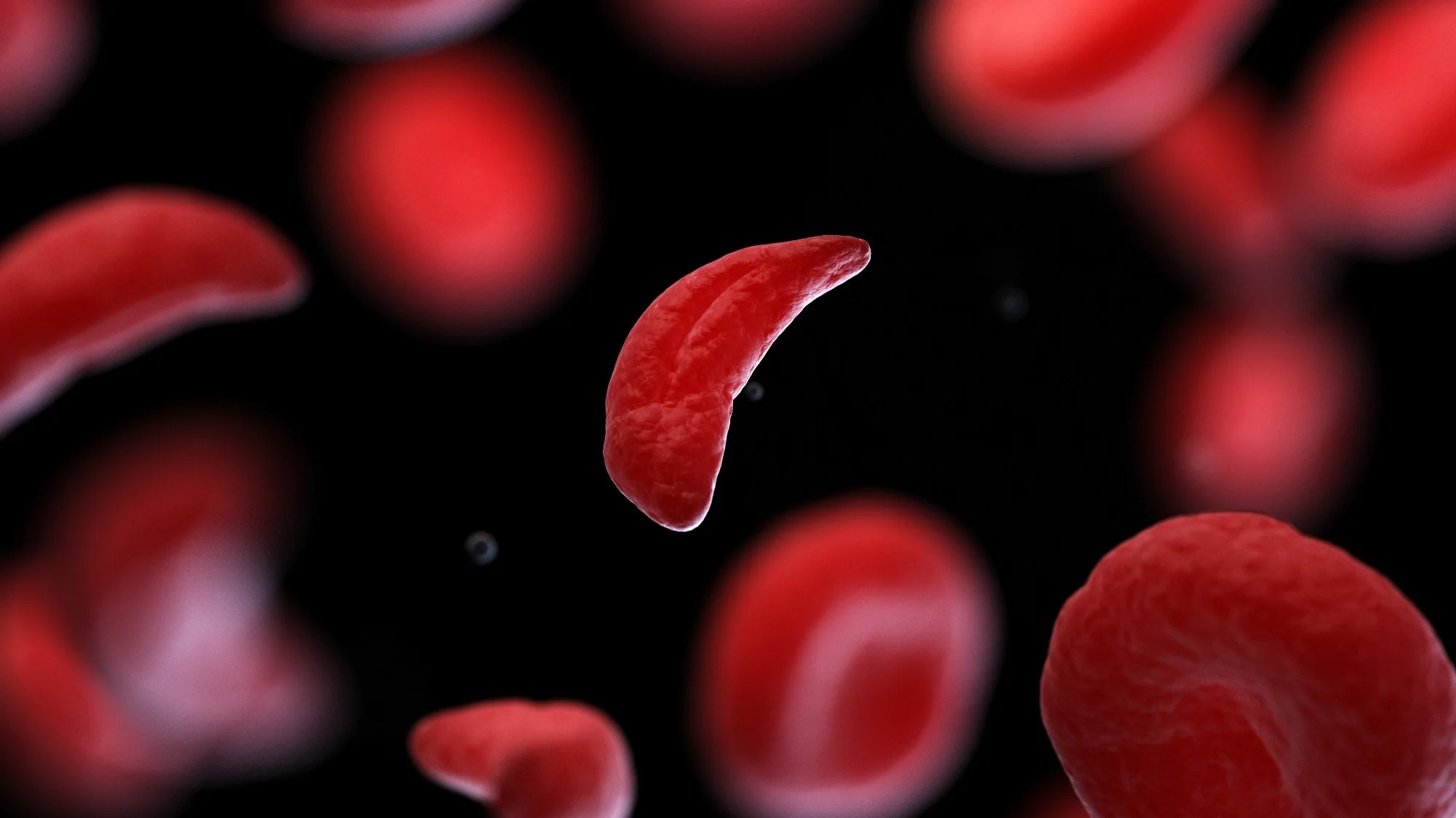
Sickle cell disease is a group of inherited disorders that affect hemoglobin, the protein in red blood cells responsible for carrying oxygen throughout the body. In SCD, abnormal hemoglobin causes red blood cells to become rigid, sticky, and crescent-shaped (like a sickle), leading to blockages in blood vessels, reduced oxygen delivery, and cell breakdown.
These misshapen cells can cause severe pain, infections, and organ damage, significantly impacting quality of life. SCD is a lifelong condition, but early diagnosis and proper management can reduce complications and improve outcomes.
Understanding the different forms of SCD and their characteristics
The most common and severe form, caused by inheriting two sickle hemoglobin genes.
A milder form caused by one sickle hemoglobin gene and one hemoglobin C gene.
Caused by one sickle hemoglobin gene and a gene for beta-thalassemia.
People affected in the United States
Babies born with SCD annually
Global cases in Africa
SCD affects approximately 100,000 people in the United States and millions globally, with an estimated 300,000 babies born with SCD annually, primarily in sub-Saharan Africa. At the national level in Uganda, it is estimated that 13.3% of individuals carry the sickle cell trait, SCD contributes 15% to the under-five mortality rate in Uganda, which is 64/1000 live births; the infant mortality rate is 43/1000 live births. Northern Uganda has the highest sickle cell prevalence of 20% specifically in the districts of Gulu, Lira, and Kitgum.
With proper care, individuals with SCD can live into their 40s or 50s, though complications can shorten lifespan, especially in low-resource settings.
About 1 in 13 African Americans carries the sickle cell trait, meaning they have one copy of the abnormal gene but do not have the disease.
SCD leads to significant healthcare costs, with annual medical expenses in the U.S. averaging $30,000 per patient due to hospitalizations and treatments.
Pain episodes, known as vaso-occlusive crises, are the hallmark of SCD and can last from hours to weeks, often requiring hospital care.
Over 75% of SCD cases occur in Africa, where limited access to healthcare exacerbates complications and mortality rates.
SCD is caused by a genetic mutation in the HBB gene, which codes for hemoglobin. The disease is inherited in an autosomal recessive pattern, meaning a child must inherit two copies of the mutated gene—one from each parent—to develop SCD.
Parents who are carriers (have sickle cell trait) have a 25% chance of passing SCD to their child with each pregnancy.
A family history of SCD or sickle cell trait increases the likelihood of inheritance.
SCD is more prevalent among people of African, Mediterranean, Middle Eastern, South Asian, Caribbean, and Latin American descent due to the historical prevalence of the sickle cell gene in malaria-endemic regions.
SCD symptoms vary in frequency and severity, often appearing in early childhood. Common symptoms include:
Early diagnosis is critical for managing SCD and preventing complications
In many countries, newborns are screened for SCD using a blood test to detect abnormal hemoglobin, allowing early intervention.
Tests like hemoglobin electrophoresis confirm the presence of sickle hemoglobin and identify the specific SCD type.
Identifies the HBB gene mutation in individuals or carriers, often used for family planning or prenatal diagnosis.
Amniocentesis or CVS can detect SCD in a fetus for at-risk pregnancies.
While there is no universal cure for SCD, various treatments can manage symptoms and improve quality of life
Over-the-counter or prescription pain relievers (e.g., ibuprofen, opioids) are used during pain crises, often with hydration and rest.
Regular transfusions increase healthy red blood cell counts, reducing stroke risk and anemia severity.
Preventive antibiotics, such as penicillin, are prescribed for children to reduce infection risk, particularly for pneumococcal infections.
This medication increases fetal hemoglobin production, reducing pain crises, hospitalizations, and the need for transfusions.
A bone marrow or stem cell transplant can cure SCD in some cases, but it is high-risk, expensive, and requires a compatible donor.
Staying hydrated, avoiding extreme temperatures, and managing stress help prevent pain crises.
SCD can lead to serious health issues if not managed properly
Blocked blood vessels in the brain increase stroke risk, particularly in children, requiring regular screening with transcranial Doppler ultrasound.
A life-threatening condition involving lung blockages, causing chest pain and difficulty breathing.
Chronic kidney disease or failure due to reduced blood flow, often requiring dialysis in severe cases.
Enlarged heart or heart failure from chronic anemia and high cardiac workload.
Retinal damage from blocked blood vessels can lead to partial or complete blindness.
High blood pressure in the lungs, increasing the risk of heart failure.
Couples with sickle cell trait can receive counseling to understand the risk of passing SCD to their children.
Blood tests identify carriers of the sickle cell trait, informing family planning decisions.
Awareness programs in high-prevalence areas promote screening and education to reduce stigma and encourage early diagnosis.
Routine vaccinations (e.g., pneumococcal, influenza) are critical for preventing infections in SCD patients.


Routine check-ups with hematologists monitor complications and adjust treatments.
A balanced diet, regular exercise (avoiding overexertion), and adequate hydration support overall health.
Counseling and support groups help address the emotional and social challenges of living with a chronic illness.
Patients and families benefit from learning about SCD and advocating for workplace or school accommodations.
SCD is a global health challenge with significant regional disparities

Home to over 75% of global SCD cases, Africa faces high mortality rates due to limited healthcare access, with many children dying before age 5. Programs like newborn screening and hydroxyurea distribution are expanding but remain underfunded.
Comprehensive care, including newborn screening and advanced treatments, has improved life expectancy, but disparities persist, with Black and Hispanic patients facing barriers to equitable care.
Countries like the UK and France have robust screening programs for at-risk populations, but awareness remains low in non-endemic regions.
SCD is prevalent in India, Saudi Arabia, and other regions, with growing efforts to implement genetic screening and public health initiatives.
Organizations like WHO and Sickle Cell Disease Association of America advocate for increased funding, research, and policy changes to address SCD worldwide, emphasizing equity in care.
Research is advancing SCD treatment and potential cures, offering hope for better outcomes
Latest advancements in gene therapy and CRISPR technology are showing promising results in clinical trials.

Clinical trials are testing therapies to insert healthy hemoglobin genes into patients' stem cells, with early successes showing reduced symptoms.
CRISPR-based technologies aim to correct the HBB gene mutation, with promising results in trials, though scalability and cost remain challenges.
Improved stem cell transplant techniques, including haploidentical (partial-match) donors, are expanding access to curative options.
Drugs like voxelotor and crizanlizumab reduce complications by improving blood flow and preventing cell adhesion, with ongoing studies to optimize their use.
Collaborative efforts, such as the Sickle In Africa consortium, are advancing research and care delivery in high-burden regions.